In our recent C&EN Webinar: Accelerating Consumer Products Reformulation with Machine Learning, we demonstrated how to leverage digital tools and technology to bring new products to market faster. The webinar was well attended by scientists, engineers, and business leaders across the product development spectrum eager to learn how these concepts can be applied to their work. We received many good questions during the event, and wanted to make our detailed responses available to the wider community. If you have any follow up questions, please reach out to us. Enthought is here to help your business navigate the challenges in R&D digital transformation and affect real change and business value generation.
Authors: Michael Heiber, Application Engineer, Materials Science Solutions Group, Chris Farrow, Vice President, Materials Science Solutions Group
(The questions below have been lightly edited for clarity.)
What courses do you recommend to take for learning how to program and develop machine learning models?
With a strong grounding in science, introductory courses in Python and Machine Learning can help you get started tackling problems like reformulation with ML. A more immersive experience with an experienced guide can get you even further faster.
Enthought can get you on the right track with our open courses. Our Applied Computing Program provides that immersive guidance for your specific challenges. We also offer a Technical Leaders Program for tackling large digital challenges with a team of Enthought Experts to help develop a digital culture in your organization.
I understand a large set of data is required for training machine learning models. For starting and generating data in the lab from experiments, what minimum amount of data do you suggest we should have to get going?
A general guideline to consider is that one needs 10-100x as many observations (measurements) as the number of features (dimensions) to develop a ‘good’ model. In some cases, far more data is needed if the observations are not reasonably well-distributed over the parameter space of interest. For example, if much of the data is clustered in a particular area of parameter space where you’ve done a lot of experiments in the past, it will be hard to generalize the model to predict behavior in other regions where you have much less data. In high-stakes decision-making situations, one might need a lot of data to feel comfortable handing over decisions to AI agents.
However, in a product R&D setting, the goal is to learn from experiments and leverage that learning to develop successful products. Innovation will frequently happen in areas where your data is likely to be sparse. In these cases, machine learning may not be able to confidently tell you how to make a successful product, but it can still be used to guide R&D efforts to make the learning process more efficient and less prone to human bias. The adaptive experimental design strategy shown in the webinar can help direct you to gather data that provides maximum benefit towards your short term design objectives and longer term research goals. With this approach, you can get started right away with only a handful of initial data.
Feature selection is also really important, and you can lean on your domain expertise to select features that you know empirically or theoretically have a large impact on the output value. This simple act of choosing features encodes some of your expert knowledge and the underlying physics and chemistry into the model and alleviates some of the data needs. Get your hands on whatever data you can and start extracting information you know is important. The tool we demonstrated during the webinar (Enthought Edge) can organize and prepare your initial dataset for machine learning.
What if the format of an experimenter’s e-notebook is not the same as another experimenter? How do you get that information into your model building tool?
Our DataOps solution, Enthought Edge, can handle importing of data from various formats into the same data model. Whatever tool you use for this, we have found that flexibility in these models is important while learning to use centralized data management and establishing data governance. The tool can help enforce structure in how you collect data, but if it does not do what scientists need, then they may not use it!
When starting with small datasets, how do you avoid overfitting?
There are a number of ways to avoid overfitting with small datasets. A simpler model with less parameters will be less prone to overfitting. In the example shown in the webinar, a relatively simple Gaussian process regression model was used without a lot of parameters. With small datasets, ML models are also likely to be more sensitive to outliers in your dataset. We performed exploratory data analysis to check for major outliers and used the median absolute percent error scoring metric which is more robust to outliers than the mean absolute percent error.
Having too many features, which are redundant or uninformative, can also lead to overfitting. For this problem, given our domain expertise, we selected only the features that we know have a significant impact on the properties of interest. If there were many more features with an unknown impact, feature selection would become an important part of the machine learning pipeline.
Finally, following best practices for cross-validation during hyperparameter optimization can also prevent this problem. In the webinar example, RepeatedKFold cross validation was used with three folds and eight repeats. With small datasets it can be more challenging to ensure that both training and validation subsets are representative of the full dataset, while also producing a validation score that is not overly sensitive to a particular random split. For this problem, the RepeatedKFold method in scikit-learn is very useful! Here is a nice Kaggle post, Dealing with Very Small Datasets, that also describes a variety of ways to prevent overfitting with small datasets.
How do you decide which samples to collect data for, and how many samples should you run? Meaning say, sample 1 (viscosity = 1, temp = 100) and sample 2 (visc = 2, temp = 200), etc.
Unlike traditional design of experiments, where you plan out a large series of experiments at the beginning and then run them all, adaptive experimental design (also commonly called Bayesian optimization or active learning) is an iterative experimentation process where you receive experiment recommendations from the machine learning-based recommendation algorithm, run a small number of experiments, process the resulting data, update your machine learning model, and then receive the next round of recommendations. This iterative process continues until you reach your objectives, exhaust your experimental budget, or reach some other predefined termination condition. Simple recommendation algorithms will specify which conditions to test depending on your design objectives and how much ‘exploration’ you want to do. For this example problem, we used the expected improvement algorithm, which takes into account both the predicted value and the uncertainty to construct a probability adjusted expected magnitude of improvement. It is one the most effective common acquisition function algorithms used for generating recommendations. These can be expanded to take into account many other factors like time, cost, and budgets.
How many experiments you run at each iteration and how many replicates you test can vary depending on the problem and on the realities of your lab process. For example, in many labs, batch processing of samples is much more efficient than making and characterizing samples one at a time. In the webinar example, we performed batches of four experiments. But, if the lab equipment is set up to efficiently run up to 12 samples at once, recommendation algorithms can be tuned to help you determine whether you should run two conditions with six replicates, three conditions with four replicates, four with three, six with two, or 12 with no replicates.
More sophisticated recommendation algorithms can also help you coordinate asynchronous parallel experimentation such as splitting up experimental tests between multiple lab scientists that may produce results at different rates. However, you don’t need to have a very advanced algorithm to get started. Even fairly basic, well-documented algorithms, like those demonstrated in the webinar, are much more efficient than the trial and error approaches that are commonly practiced in the lab.
Is 25% model accuracy typical for such approaches, or can we further improve this accuracy?
In the webinar example, the models for predicting the application viscosity and the yield stress both produced a final scoring metric of 25% median absolute percent error from the outer loop of our nested cross-validation. This score provides a best estimate of how the model will perform relative to new data that the model has not seen yet. For similar types of problems, we’ve seen model accuracies from down around 10% up to 50%–it can vary a lot depending on the details of the behavior you are trying to predict and the dataset at hand. Whether this amount of accuracy is ‘good enough’ to be useful really depends on the problem and the business constraints. For the design problem presented in the webinar, it is good enough to provide experiment recommendations that direct us toward meeting our design objectives and should be much more effective than a grid search over the parameter space or a trial and error approach.
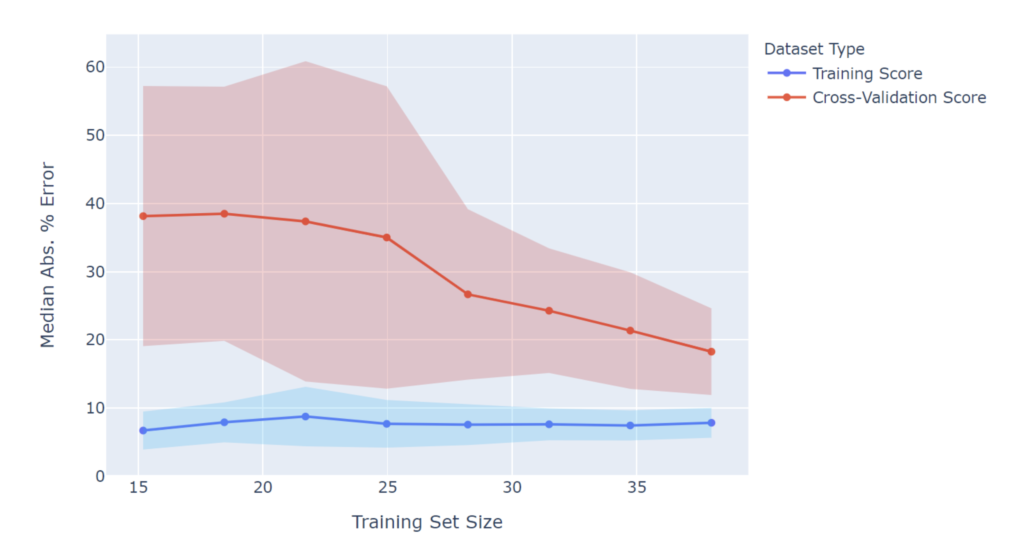
Nevertheless, a better performing model would help, and it’s useful to understand why model performance doesn’t meet initial expectations. One common way to diagnose model performance is to use a learning curve. Analysis of the learning curve can indicate whether one can expect the model performance to continue improving as more data is gathered. For example, let’s take a closer look at the learning curve for the viscosity model. Note that in this figure a lower score is better. Performance on the training set is very good at ~8%, regardless of the training set size. However, the cross-validation score continually decreases as the training set size increases, and even with all of the data included, has not yet converged. This indicates that gathering more data would likely lead to further improvements in model performance and reach a score closer to the ~8% metric obtained on the training data. With more data, a final viscosity model score of 10-15% median absolute percent error seems possible.
Viscosity Learning Curve
In your example, you used a Gaussian process regression model. Have you tried other popular machine learning models such as neural networks?
For this kind of product or material property optimization problem, one should use a model that generates both predictions and prediction uncertainties, in order to drive the recommendation system. Most machine learning models do not do this, which is why Gaussian process regression is the most common method for the adaptive experimental design approach described here. However, there are other methods that are sometimes used. One alternative is random forest regression. While standard random forest models do not produce prediction uncertainty, this can be estimated using various methods. For example, the scikit-garden package implements a random forest quantile regressor that produces uncertainty estimates. Bayesian neural networks can also potentially be used for this purpose but it is not common. In most cases, Gaussian process regression works well and is a good default starting point.
Can an existing, well-established model be updated to incorporate new design criteria?
Yes. In the webinar, we showed how the machine learning model is re-trained after each batch of new data is gathered. We can continue this process until we reach our design objectives. Eventually, though, we’ll have a new design problem to create a new yield stress fluid product with different design criteria. If we still have the same tunable parameters, then we can use the same machine learning model for the new design problem. As we learn about new independent variables that we can use to tune product performance, the machine learning model must be expanded to learn these relationships by adding the new variables as features of the model. For example, perhaps we want to try changing the polymer used in the formulation. To teach the model the effect of this choice, we need to add appropriate features that capture the important changes being made, such as molecular weight, branching ratio, chemical fingerprints, and other descriptors.
I am interested in how you see this approach fit into food R&D, as food acceptance is a huge piece of the puzzle. Do you believe we can use models to predict acceptance, and how much more challenging is that?
The data management and adaptive experimental design approach discussed in the webinar should fit into food R&D or any other complex product development area. If food acceptance is a binary metric (pass or fail), then perhaps the problem is best simplified to be a classification problem. There are many different classification models one could use, but one is the Gaussian process classification model, which would provide probabilistic predictions of pass or fail. Alternatively, if your food products are graded on a numerical scale, a Gaussian process regression model could potentially be used here as well.
As with most problems in this area, the largest challenges are often centered around the data, not the machine learning models. Developing an efficient process for generating high quality data and making sure that it is properly organized and accessible for machine learning development and use is usually more of a barrier than the machine learning model development itself. When high quality data is hard to come by and expensive to generate, it can also help to break down big prediction tasks and decisions into smaller ones where you have more data, or where you have identified critical bottlenecks in your product development process.
Is machine learning applicable to complex polymerization systems? For instance, creating a model for two phases since mathematical models will not reflect it properly.
Yes. Data-driven methods are especially useful for predicting the outcomes of complex processes where there are no existing well-performing theoretical or empirical models. Most machine learning models are non-parametric, which means that you don’t need to know the functional form of the behavior of interest. For example, the Gaussian process regression model used in the webinar can fit a wide variety of functional forms as long as they are smooth and continuous. For behavior that is discontinuous, other models like random forest regression may be more appropriate.
Related Content
Digital Transformation vs. Digital Enhancement: A Starting Decision Framework for Technology Initiatives in R&D
Leveraging advanced technology like generative AI through digital transformation (not digital enhancement) is how to get the biggest returns in scientific R&D.
Digital Transformation in Practice
There is much more to digital transformation than technology, and a holistic strategy is crucial for the journey.
Leveraging AI for More Efficient Research in BioPharma
In the rapidly-evolving landscape of drug discovery and development, traditional approaches to R&D in biopharma are no longer sufficient. Artificial intelligence (AI) continues to be a...
Utilizing LLMs Today in Industrial Materials and Chemical R&D
Leveraging large language models (LLMs) in materials science and chemical R&D isn't just a speculative venture for some AI future. There are two primary use...
Top 10 AI Concepts Every Scientific R&D Leader Should Know
R&D leaders and scientists need a working understanding of key AI concepts so they can more effectively develop future-forward data strategies and lead the charge...
Why A Data Fabric is Essential for Modern R&D
Scattered and siloed data is one of the top challenges slowing down scientific discovery and innovation today. What every R&D organization needs is a data...
Top 5 Takeaways from the American Chemical Society (ACS) 2023 Fall Meeting: R&D Data, Generative AI and More
By Mike Heiber, Ph.D., Materials Informatics Manager Enthought, Materials Science Solutions The American Chemical Society (ACS) is a premier scientific organization with members all over…
Real Scientists Make Their Own Tools
There’s a long history of scientists who built new tools to enable their discoveries. Tycho Brahe built a quadrant that allowed him to observe the…
How IT Contributes to Successful Science
With the increasing importance of AI and machine learning in science and engineering, it is critical that the leadership of R&D and IT groups at...
From Data to Discovery: Exploring the Potential of Generative Models in Materials Informatics Solutions
Generative models can be used in many more areas than just language generation, with one particularly promising area: molecule generation for chemical product development.